はじめに
こんにちは。TIG/DXチームの伊藤です。GCP連載の4日目です。
さて、私は業務ではGCPとTerraformを扱っています。コンピューティング・ネットワーキングなどが現在の業務領域でである一方で、ビッグデータ・機械学習関連のサービスはあまり使う機会がなく理解もまだまだな部分が多いです。今回は、Cloud Life Sciencesという、ビッグデータの中でも毛色の違うサービスを調べてみようと思いこの連載に参加しております。割と読み物に近くなったのでさらっと読み流していただければ幸いです。
私の今まで
本記事を書くためのモチベーションとしては昔話をすこし挟みます。現在はインフラやネットワークエンジニアとして日々働いていますが、大学時代は化学を専攻し大学院では感染症を専門に研究していました。最終的に遺伝子を触ることなく修了しましたが、生物系に少しでも在籍していたこともあり、今回はわずかな記憶を頼りながら調べています。この記事が最終的にかつての研究の何かに使えたかもしれないと私の中で知識が落とし込めればいいなと考えています。
Cloud Life Sciencesについて

GCPの中ではビッグデータに属するサービスになります。大規模な生物医学データの処理を費用対効果が高くできることがウリで、対象ユーザーはライフ サイエンス機関と学術研究機関向けとなっています。特に強く謳われているのが、研究に集中できることと結果の再現性にです。遺伝子情報は特に大きなデータ量になるので、得たデータの検証をスピードアップさせることは研究スピード全体を上げることにも繋がります。
また、アーキテクチャは以下になっており、サービス単体としてではなく、1つのプラットフォームとして動くようです。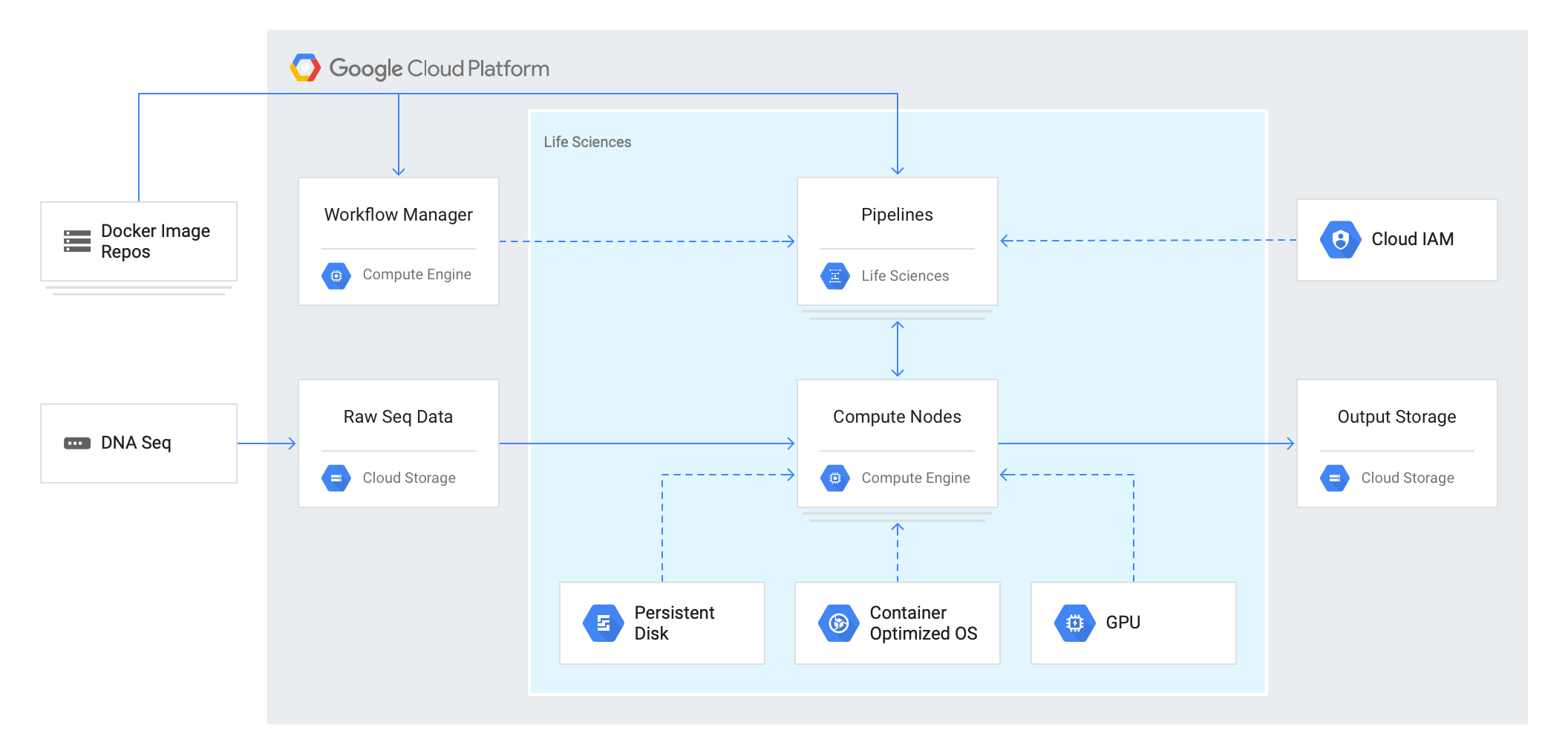
(引用:https://cloud.google.com/life-sciences/?hl=ja)
VCF
Variant Call Formatと言われる、遺伝子配列の変異データを保存するときに利用されるファイル形式になります。ファイル内の記載内容は以下になります。
- VCFヘッダー行(1~8行目)
- データのフォーマット行(カラムタイトル)
- 染色体番号
- 塩基の位置
- 塩基(変異箇所)のID
- リファレンス配列上の塩基
- シーケンスデータ上の塩基
- シーケンシングデータのスコア
- フィルタリング条件の通過の有無
- key:dataの形の追加情報
このような形でシーケンス結果を記載しています。DNAの塩基配列についてはアデニン(A)、チミン(T)、グアニン(G)、シトシン(C)の4種類がありますが、それぞれ、A=T、G≡Cとしてペアになっているのでぜひ試すときには思い出してみてください。また後のクエリに使います。
Variant Transformsツール
こちらのツールはGoogleが作成したオープンソースになります。実際にBigQueryに読み込ませるためにはデータの加工が必要になりますので、こちらのツールを使って分析に最適な形で出力できます。実際のフローにはDataflowが使用されており、BigQueryで分析するまでは以下の図になります。
実際に流れを掴む
実際に公開データセットを使ってバリアントを使って少しいじってみましょう。ここは実際のチュートリアルの一部を行なっているので、本記事以外の方法を試したい方はこちらから確認してください。Cloud Consoleで行うとクリーンに進められます。
GCSからVariant TransformsツールからBigQueryに送る
GCSにバリアントファイルを配置したら実際にツールを使います。使うためには公式のDockerイメージがあるのでこちらを落としてきます。イメージが2GB近くあるので、ダウンロードは気長に待ちましょう。
docker pull gcr.io/cloud-lifesciences/gcp-variant-transforms |
次にスクリプトを実行します。変数は実際に使っているプロジェクト名、バケットなどに置き換えてください。
|
スクリプト実行後は時間がかかるようなので、これも気長に待ちます。
実行が終わったらbqコマンドでアウトプットがあるか確認しましょう。
bq ls --format=pretty GOOGLE_CLOUD_PROJECT:BIGQUERY_DATASET |
これでVariant Transformツールを使ってファイルをBigQueryに適した形にすることが出来ました。
BigQueryに読み込ませる
こちらでは公開プロジェクトのデータを使ってバリアントの分析を行います。データを用いるプロジェクトはIllumina Platinum Genomes、テーブル名はplatinum_genomes_deepvariant_variants_20180823になります。実際のテーブルを5件出力しました。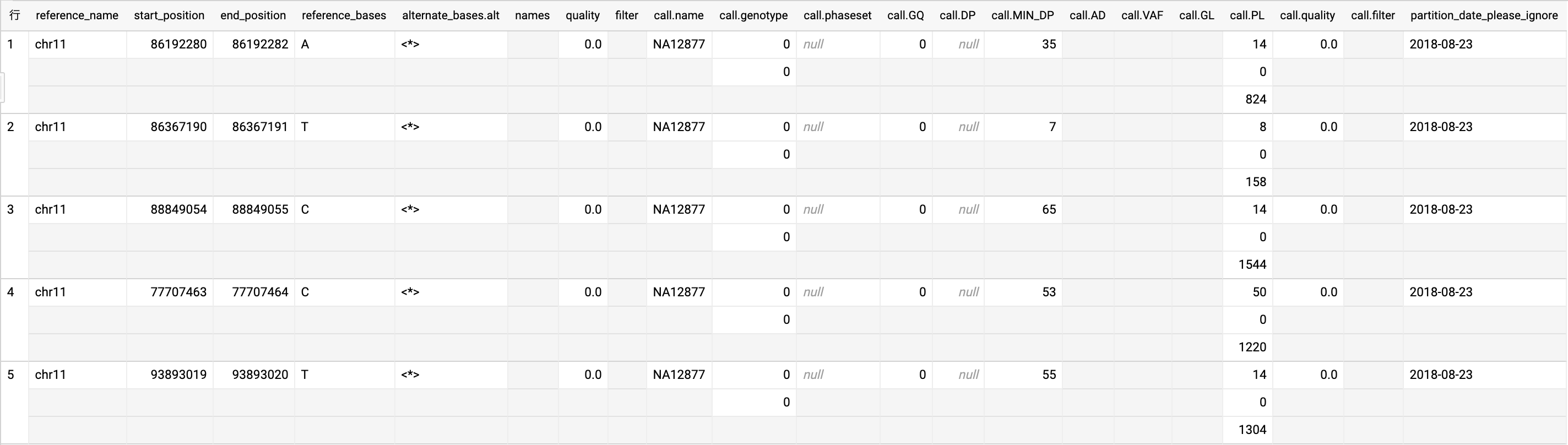
実際に流すクエリは以下になります。
WITH filtered_snp_calls AS ( |
mutation_type_countsのところで、正確にペアになっている塩基対(A=T、もしくはG≡C)についてはtransitions、正しいペアになっていない塩基対はtransversions(変異)しているものをそれぞれint型にCASTして合計を取っています。上記クエリをBigQueryに流すと以下の結果が出力されます(上記10件)。
| 行 | referencce_name | name | transitions | transversions | titv |
|---|---|---|---|---|---|
| 1 | chr22 | NA12892 | 35299 | 15017 | 2.3506026503296265 |
| 2 | chr22 | NA12889 | 34091 | 14624 | 2.331167943107221 |
| 3 | chr17 | NA12892 | 67297 | 28885 | 2.3298251687727194 |
| 4 | chr22 | NA12878 | 33627 | 14439 | 2.3289008934136715 |
| 5 | chr22 | NA12877 | 34751 | 14956 | 2.3235490772933938 |
| 6 | chr22 | NA12891 | 33534 | 14434 | 2.323264514341139 |
| 7 | chr17 | NA12877 | 70600 | 30404 | 2.3220628864623074 |
| 8 | chr17 | NA12878 | 66010 | 28475 | 2.3181738366988585 |
| 9 | chr17 | NA12890 | 67242 | 29057 | 2.314141170802216 |
| 10 | chr17 | NA12889 | 69767 | 30189 | 2.311007320547219 |
最終列のtitvでは正しいペア、および変異の入ったペアの比をとっているので、各リファレンスに対してどれくらい変異は入っているかがわかります。ここでは20 GBほど読み込んでいますが、実際にクエリにかかった時間は1秒を切っているので、ローカルで同様のクエリ実行するよりははるかに早いでしょう。
まとめ
本記事では簡単ですが、Cloud Life Sciencesを触る記事を書いてきました。エンジニアとして分析に使うものはうっすら理解していても、実際に使うファイルフォーマットなどは知識が深くないとそもそも何もできないなと改めて感じたところです。今回はVCFファイルという専門的なファイルからBigQueryに流すところまで行いましたが、Cloud Life Sciencesはもともと Google Genomicsというサービスだったようで、以前よりGoogleが力を入れていた部分の1つであることが今回色々調べてわかりました。医療分野は正確さを求められる一方で、そのデータ量は膨大な量になります。大学院時代に肌で感じたことですが、ローカルで分析している先輩がかなり時間をかけていたことを思い出しました。このサービスの知名度が上がるとともに、研究ユース、非エンジニアでも使いやすいプラットフォームになるとより良いなと思いました。
GCP連載の4日目でした。次は市川さんの【もう鍵なくさない】GCPのSecret ManagerとBerglasで幸せになるです。
最後に
先日、GCPのProfessional Cloud Architectに合格しました! 結果が出力される瞬間はなかなかドキドキでしたが、なんとか合格できました。私も参考にしましたが、本技術ブログでも合格体験記を出しておりますので、そちらもぜひご覧ください!
【合格記】GCP Professional Cloud Architect認定資格を振り返る