はじめに
こんにちは。SAIG所属の農見と申します。
この度、研究で用いたB細胞エピトープ予測用データセットとサンプルコードを公開したので、その解説をしようと思います。(これのプレスリリース)
データセットについて
簡単に言うと、B細胞エピトープが分かればワクチン作成の大きな助けになる、だからCOVID-19の原因ウィルスであるSARS-CoV-2が持つタンパク質のB細胞エピトープを予測して、ワクチン開発の役に立てようぜというデータセットです。
B細胞エピトープについてもう少し詳しく知りたい方は我々の論文を読むか免疫学の本を読んでみるといいと思います。
サンプルコードについて
では本題のサンプルコードの解説をしていきます。
INPUT_DIR = '../input/epitope-prediction'
bcell = pd.read_csv(f'{INPUT_DIR}/input_bcell.csv')
sars = pd.read_csv(f'{INPUT_DIR}/input_sars.csv')
covid = pd.read_csv(f'{INPUT_DIR}/input_covid.csv')
bcell_sars = pd.concat([bcell, sars], axis=0, ignore_index=True)
bcell_sars.head()
|
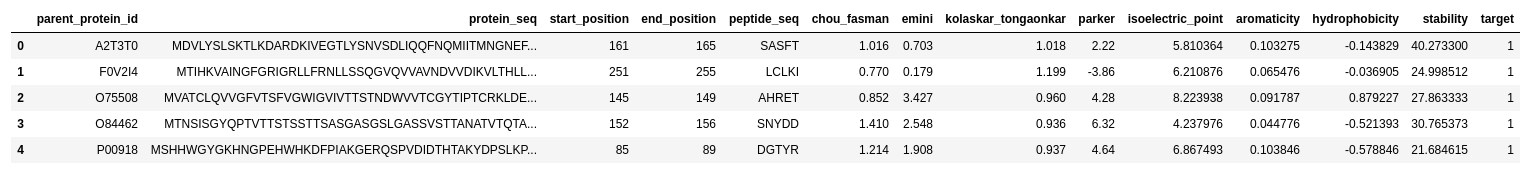
最初の部分でデータがどんなものなのかということを見ています。基本中の基本ですね、データがどんなものなのかということが分からないと方針の立てようがないです。
各カラムは
parent_protein_id : タンパク質のUniProt IDprotein_seq : タンパク質の配列start_position : エピトープの開始位置end_position : エピトープの終了位置peptide_seq : エピトープ配列chou_fasman : エピトープ特徴量, $\beta$ ターンemini : エピトープ特徴量, relative surface accessibilitykolaskar_tongaonkar : エピトープ特徴量, 抗原性parker : エピトープ特徴量, 疎水性isoelectric_point : タンパク質特徴量,等電点aromacity: タンパク質特徴量,芳香族アミノ酸の割合hydrophobicity : タンパク質特徴量,疎水性stability : タンパク質特徴量,安定性target : 抗体価
となっています。予測したいものはtarget(抗体価)でこれは0と1のbinaryの値を取ることが分かります。
np.sum(bcell_sars.isnull())
|
また、欠損値があるかどうかのチェックもしました。幸いこのデータには欠損値はないので、そのままデータを使うことが出来ます。ということで早速B-cellのデータを利用してCOVID-19に近いとされるSARSのBcellエピトープ予測を行いました。
Task1 : SARS予測
Bcellデータセットのみを使用してSARSデータセットの抗体価を予測するタスク。
SARS-CoVとSARS-CoV-2は配列に類似性があるためTask1ではSARSの予測を行っている。
for df in [bcell, sars, covid, bcell_sars]:
df["length"] = df["end_position"] - df["start_position"] + 1
kf = GroupKFold(n_splits = 5)
oof = np.zeros(len(bcell))
preds = np.zeros(len(sars))
feature_importance = pd.DataFrame()
feature_columns = ["chou_fasman","emini","kolaskar_tongaonkar","parker","length","isoelectric_point","aromaticity","hydrophobicity","stability"]
target = "target"
for i,(train_index,valid_index) in enumerate(kf.split(bcell[feature_columns],bcell["target"],bcell["parent_protein_id"])):
train_x = bcell.loc[train_index][feature_columns].values
train_y = bcell.loc[train_index][target].values
valid_x = bcell.loc[valid_index][feature_columns].values
valid_y = bcell.loc[valid_index][target].values
train_data = lgb.Dataset(train_x, label = train_y)
valid_data = lgb.Dataset(valid_x, label = valid_y)
model = lgb.train(params, train_data, valid_sets = [valid_data], verbose_eval=20,num_boost_round=500,early_stopping_rounds=50)
oof[valid_index] = model.predict(valid_x)
preds += model.predict(sars[feature_columns].values)/kf.n_splits
feature_importance["feature"] = feature_columns
feature_importance["importance"+str(i)] = model.feature_importance()
roc_auc_score(bcell["target"],oof)
|
lengthという特徴量を作成して、普通に5-fold cross validationしているだけですが、注目すべき部分はGroupKFoldを使っている点です。ただ、GroupKFoldを知らない人もいると思うので先にGroupKFoldの説明をします。
https://scikit-learn.org/stable/auto_examples/model_selection/plot_cv_indices.html#sphx-glr-auto-examples-model-selection-plot-cv-indices-pyから引用。
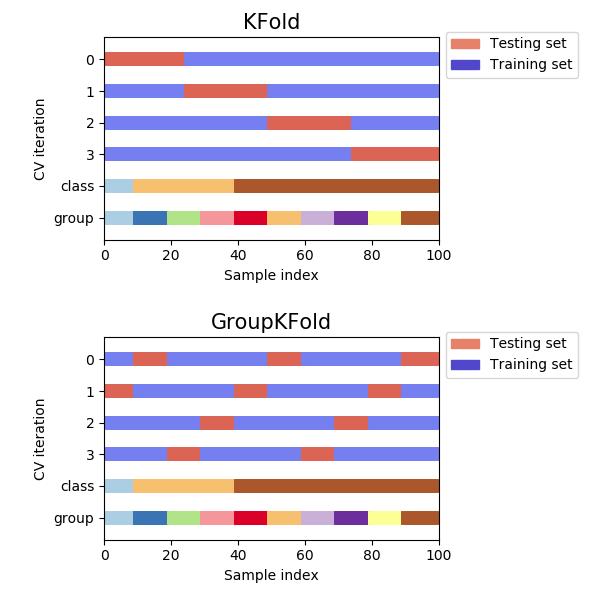
これを見てみるとGroupKFoldはKFoldと異なりGroupをまたぐことのない分け方をしていることが分かります。
で、今回何故parent_protein_idをgroupとしてGroupKFoldをしているかというとタンパク質ごとに分けないと不当に精度が高くなる恐れがあるためです。これは単純な理由でtargetに1しかないタンパク質や0しかないタンパク質というものが存在します。これをtrainとvalidationに混在させてしまうとより精度が高くなるというのは想像に難くないと思います。
select = [i != "feature" for i in feature_importance.columns]
select = feature_importance.columns[select]
feature_importance[select] = feature_importance[select]/feature_importance[select].sum()
feature_importance["importance"] = feature_importance.select_dtypes(include=[np.number]).mean(axis=1)
sns.barplot(x="importance", y="feature", data=feature_importance.sort_values(by="importance", ascending=False));
plt.title('GBDT Features (avg over folds)');
|
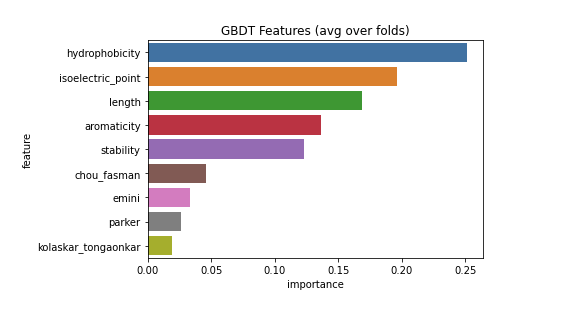
LightGBMではFeature importanceという指標で、どの特徴量がより予測に寄与したのかを見ることが出来ます。
今回の例で見ると予測に一番重要だったのはhydrophobicity(タンパク質の疎水性)であることが分かります。
またSARSは正解データがあるので予測がどれだけあたっていたかも見てみましょう。
from sklearn.metrics import accuracy_score, classification_report
print("Accuracy:", accuracy_score(sars["target"].values, np.int32(preds >= 0.5)))
print(classification_report(sars["target"].values, np.int32(preds >= 0.5)))
|
Accuracy: 0.7403846153846154
precision recall f1-score support
0 0.74 0.99 0.85 380
1 0.78 0.05 0.09 140
accuracy 0.74 520
macro avg 0.76 0.52 0.47 520
weighted avg 0.75 0.74 0.65 520
|
これを見ると1に対するrecallがとても低く改善の余地があることが分かります。
Task2 : SARS-CoV-2 (COVID-19) 予測
BcellとSARSのデータセットを利用して、SARS-CoV-2の抗体価を予測するタスク。
SARS-CoV-2には抗体価データがないところが難しいポイントです。
kf = GroupKFold(n_splits = 5)
oof = np.zeros(len(bcell_sars))
preds = np.zeros(len(covid))
feature_importance = pd.DataFrame()
feature_columns = ["chou_fasman","emini","kolaskar_tongaonkar","parker","length","isoelectric_point","aromaticity","hydrophobicity","stability"]
target = "target"
for i,(train_index,valid_index) in enumerate(kf.split(bcell_sars[feature_columns],bcell_sars["target"],bcell_sars["parent_protein_id"])):
train_x = bcell_sars.loc[train_index][feature_columns].values
train_y = bcell_sars.loc[train_index][target].values
valid_x = bcell_sars.loc[valid_index][feature_columns].values
valid_y = bcell_sars.loc[valid_index][target].values
train_data = lgb.Dataset(train_x, label = train_y)
valid_data = lgb.Dataset(valid_x, label = valid_y)
model = lgb.train(params, train_data, valid_sets = [valid_data], verbose_eval=20,num_boost_round=500,early_stopping_rounds=50)
oof[valid_index] = model.predict(valid_x)
preds += model.predict(covid[feature_columns].values)/kf.n_splits
feature_importance["feature"] = feature_columns
feature_importance["importance"+str(i)] = model.feature_importance()
roc_auc_score(bcell_sars["target"],oof)
covid["target"] = preds
covid.to_csv("sub.csv",index=False)
|
SARS-CoV-2への予測も同様にして予測結果を出力しますが、正解データがないので良し悪しの判断は難しいです。この部分は今後の生体内での実験で得られるB細胞エピトープの情報を踏まえた上で判断することになると思います。
まとめ
今回のデータセット、ソースコード、論文の公開により、医学の知見を持たないAI研究者でも容易にワクチン開発用データで実験することが出来るようになりました。このデータセットを使用して得られた知見をKaggle notebook等で公開していただけると幸いです。